Earlier this summer the U.S. Food and Drug Administration (FDA) issued draft guidance regarding patient-matched guides to orthopedic implants.
FDA’s Draft Guidance for Patient-Matched Ortho Implant Guides
2 min read Premium comments
#fdaSecondary#draftguidance#usfoodanddrugadministration
In the draft guidance, the FDA “provides recommendations regarding information that should be included in regulatory submissions for patient-matched guides to orthopedic implants.” It also provides “recommendations that manufacturers should consider when developing their design process for these device types.”
Draft guidance, when finalized, represents the current thinking of the FDA. It does not establish rights and is not binding.
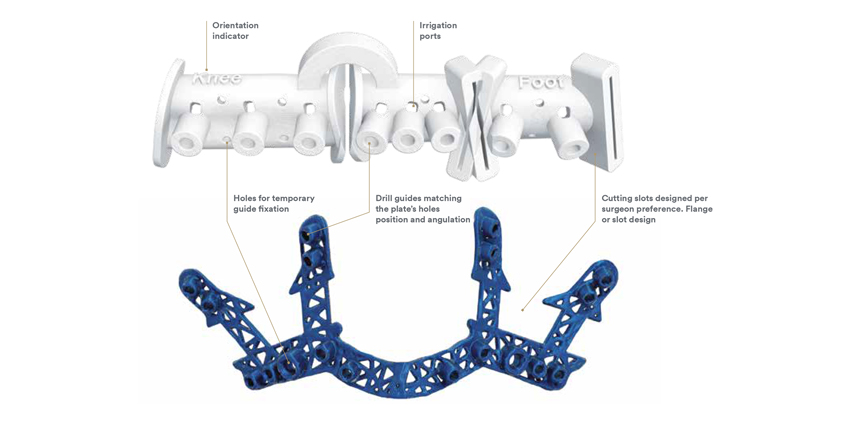
As discussed in the draft guidance, patient-matched guides are distinct from custom devices. Here, patient-matched guides are “designed to implement, in part or in whole, the pre-operative plan concurred upon by the patient’s healthcare professional.” The FDA limits the draft guidance to patient-matched guides intended for use with “legally marketed orthopedic implant systems that include recommended alignment parameters relative to rigid anatomical structures that can be identified on pre-operative imaging.”
The FDA provides guidance for the patient-matched guides indications for use. The FDA provides a list of the indications for use of an orthopedic patient-matched guide in the draft guidance.
The FDA also provides guidance regarding device description, indicating that the device description “should encompass the patient-matched guide design as well as the design process and surgical use.” The FDA details the following which should be included in a complete device description:
- Patient-matched guide description
- General design process description
- Patient image acquisition description
- Image quality control, segmentation, and anatomical definitions description
- Pre-operative planning and healthcare professional concurrence description
- Guide design and patient-matched features definition description
- Guide construction description
- Surgical technique description
The FDA also touched on software used in the development of patient-matched guides. It refers manufacturers to existing software guidance, “Content of Premarket Submissions for Device Software Functions.”
The FDA also discusses when modifications may necessitate a new 510(k) submission. It details a non-exhaustive list that includes four changes as requiring a new 510(k) submission and refers readers to additional FDA guidance for more details including the following: “Deciding When to Submit a 510(k) for a Change to an Existing Device” and “Deciding When to Submit a 510(k) for a Software Change to an Existing Device.”
React:
Discussion
This is a fascinating development. In my practice we've seen similar outcomes with the revised protocol. The key differentiator seems to be patient selection criteria. Has anyone else noticed the correlation with BMI thresholds?
Great point. I'd push back slightly on the conclusion, the sample size in the cited study is too small to draw population-level inferences. That said, the directional signal is compelling and worth a larger RCT.
We implemented a similar approach last year. Early results are promising but we're still gathering 12-month follow-up data. Happy to share our protocol if anyone is interested.
Join the conversation
Orthopedic professionals are discussing this. Sign in and upgrade to read every comment and add your voice.