The FDA’s Orthopaedic & Rehabilitation Devices panel is planning to meet on December 12, 2014 to consider VertiFlex Inc.’s pre-market approval application (PMA) for the company’s Superion ISS (interspinous spacer).
FDA Ortho Panel to Review VertiFlex Spine Device
2 min read Premium comments
Secondary
Compared to X-STOP
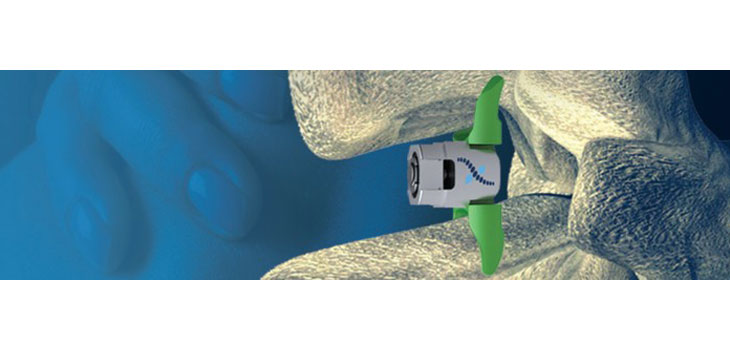
Company President and CEO Earl Fender says the panel is going to see evidence from the “largest and most rigorous FDA trial ever completed for spinal stenosis.” The device is for the treatment of patients with lumbar spinal stenosis. The IDE (investigational device exemption) trial involved enrollments of 470 patients between June 2008 and December 2012 at 31 centers across the U.S. Patients were randomized 1:1 to either the Superion ISS or the commercially available X-STOP IPD.
The company submitted its final module of the PMA this past April. The final module covers the clinical results from the Superion IDE trial evaluating the device’s safety and effectiveness.
Design Goal
The device is designed to achieve indirect spinal decompression for patients suffering from neurogenic intermittent claudication due to moderate lumbar spinal stenosis. Superion is implanted minimally invasively through a cannula designed to be less traumatic to the patient. The Superion ISS can be implanted under general or local anesthesia.
Proposed Indications
The proposed “Indication for Use” for the device, as stated in the PMA, is as follows:
“Treat skeletally mature patients suffering from pain, numbness, and/or cramping in the legs (neurogenic intermittent claudication) secondary to a diagnosis of moderate lumbar spinal stenosis, with or without Grade 1 spondylolisthesis, confirmed by X-ray, MRI and/or CT evidence of thickened ligamentum flavum, narrowed lateral recess, and/or central canal or foraminal narrowing.”
“Patients with impaired physical function who experience relief in flexion from symptoms of leg/buttock/groin pain, numbness, and/or cramping, with or without back pain. The Superion ISS may be implanted at one or two adjacent lumbar (L) levels in patients in whom treatment is indicated at no more than two levels, from L1 to L5.”
The panel will meet on December 12, 2014, from 8:00 a.m. to 6:00 p.m. at the Holiday Inn Washington-College Park, 10000 Baltimore Avenue, College Park, Maryland.
VertiFlex, Inc.
The company, founded in 2005 and headquartered in San Clemente, California, has developed proprietary, minimally invasive technologies for performing both indirect and direct decompressions of the lumbar spine. These procedures, according to the company, fill the gap in the stenosis treatment continuum between conservative care and traditional spine surgery, providing new options for interventional spine physicians and less invasive options for traditional spine surgeons.
The FDA cleared the company’s Totalis direct compression system and UniVise spinous process fixation device in January 2013. A year later, CMS granted coverage for the compression system.
React:
Discussion
This is a fascinating development. In my practice we've seen similar outcomes with the revised protocol. The key differentiator seems to be patient selection criteria. Has anyone else noticed the correlation with BMI thresholds?
Great point. I'd push back slightly on the conclusion, the sample size in the cited study is too small to draw population-level inferences. That said, the directional signal is compelling and worth a larger RCT.
We implemented a similar approach last year. Early results are promising but we're still gathering 12-month follow-up data. Happy to share our protocol if anyone is interested.
Join the conversation
Orthopedic professionals are discussing this. Sign in and upgrade to read every comment and add your voice.