Imagine the massive effort required to spend a full day presenting evidence to the FDA orthopedic panel to prove your device is safe and effective. SpinalMotion, Inc. is doubling down and going for two days in July.
SpinalMotion’s Kineflex to FDA Panel in July
2 min read Premium comments
Secondary
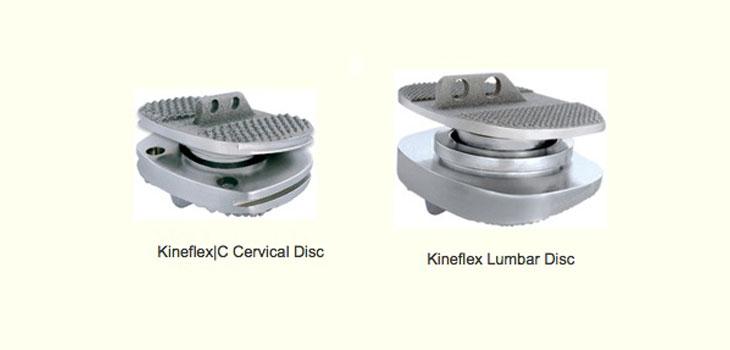
The company is going to the panel on July 24 and 25, 2013, to present evidence of the safety and effectiveness of the Kineflex/C Cervical and the Kineflex Lumbar Artificial Discs.
The Kineflex/C is a metal-on-metal (cobalt chrome molybdenum alloy) cervical total disc replacement device. The device is indicated for reconstruction of the intervertebral disc at one level from C3-C7 following single-level discectomy for intractable radiculopathy or myelopathy due to a single-level abnormality localized to the disc space.
The Kineflex Lumbar disc is a metal-on-metal (cobalt chrome molybdenum alloy) lumbar total disc replacement device. The device is indicated for reconstruction of the intervertebral disc at one level (L4-L5 or L5-S1) following single-level discectomy for lumbar degenerative disc disease (DDD) where DDD is defined as discogenic back pain with degeneration of the disc as confirmed by patient history, physical examination, and radiographic studies.
Company President and CEO David Hovda told us he can’t say much before the meeting but is looking forward to sharing the company’s long term, positive clinical data with the panel.
The company received IDE (Investigation Device Exemption) approvals from the FDA in 2005 to commence studies. The cervical trial involves over 20 U.S. sites. The lumbar trial also involves over 20 U.S. sites, and is a randomized study comparing the disc to another FDA approved lumbar artificial disc. Both trials require a two-year follow-up period.
In June 2007 the company completed enrollment of its cervical clinical trial. Enrollment in the company’s first IDE clinical trial, evaluating the lumbar disc was completed in 2006.
On March 20, 2012 the company announced that it received CE Mark for two sub-5mm Kineflex/C cervical total disc replacements.
The company conducted the IDE clinical study of the cervical disc compared to anterior cervical discectomy and fusion in the U.S., and submitted its premarket approval (PMA) application in 2010. A cervical disc paper was selected for the Best Papers section at the 2010 North American Spine Society’s Annual Meeting.
In October 2010 the company announced it completed enrollment in an international clinical study evaluating the lumbar disc inserted via a minimally invasive lateral approach.
The meeting will be held at the Hilton Washington DC North/Gaithersburg on 620 Perry Parkway in Gaithersburg, Maryland. The FDA intends to make background material available to the public no later than two business days before the meeting.
React:
Discussion
This is a fascinating development. In my practice we've seen similar outcomes with the revised protocol. The key differentiator seems to be patient selection criteria. Has anyone else noticed the correlation with BMI thresholds?
Great point. I'd push back slightly on the conclusion, the sample size in the cited study is too small to draw population-level inferences. That said, the directional signal is compelling and worth a larger RCT.
We implemented a similar approach last year. Early results are promising but we're still gathering 12-month follow-up data. Happy to share our protocol if anyone is interested.
Join the conversation
Orthopedic professionals are discussing this. Sign in and upgrade to read every comment and add your voice.